The Visible Alpha GLP-1 Drug Monitor tracks the current and projected growth of the GLP-1 (glucagon-like peptide-1) receptor agonist family of therapeutics marketed by publicly traded companies for type 2 diabetes, obesity or weight management, and other indications. The monitor evaluates both approved GLP-1 drugs and developing GLP-1 pathway-related drug candidates in the pipeline based on revenue potential while taking into account innovation and mechanism-of-action impact as well as clinical and regulatory risk. This update follows the first GLP-1 monitor published on March 26, 2024.
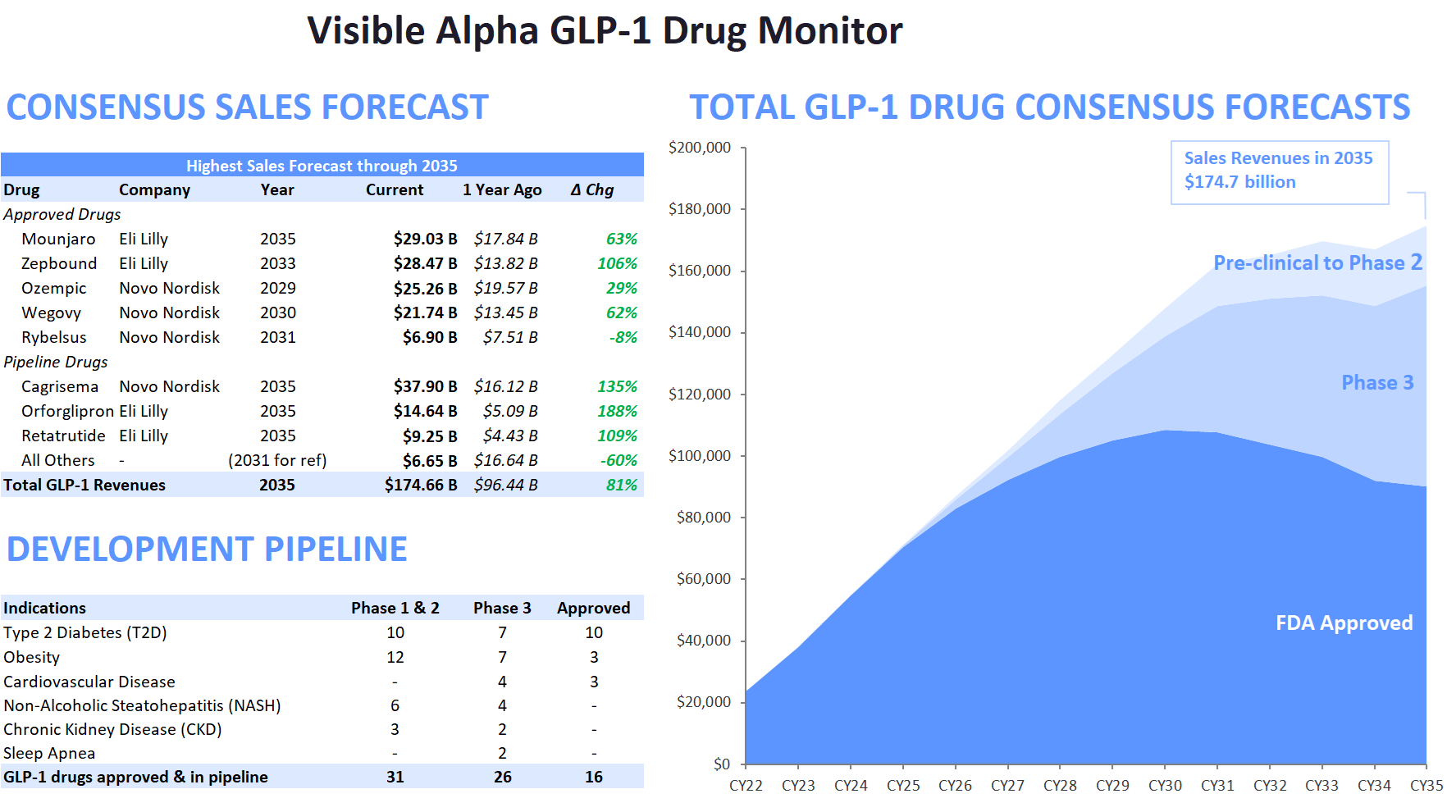
Figure 1: The Visible Alpha GLP-1 Drug Monitor
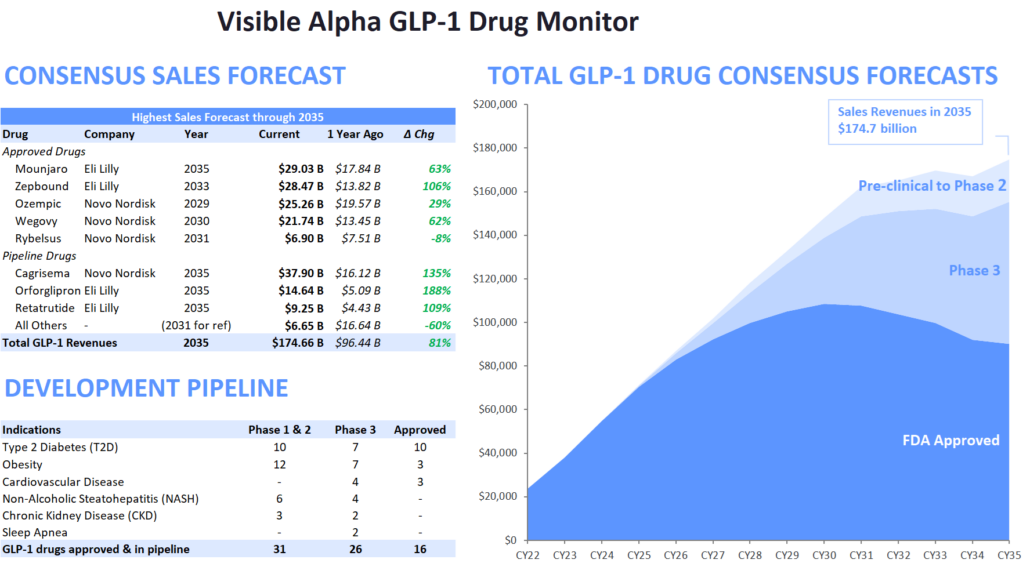
(Change from the previous GLP1 Monitor – Pfizer’s Danuglipiron, in Phase2, has been removed from pipeline drug candidates, to depict only lead Phase 3 drug candidates) Source: Visible Alpha BioPharma consensus (March 22, 2024); forecasts are in millions of U.S. dollars
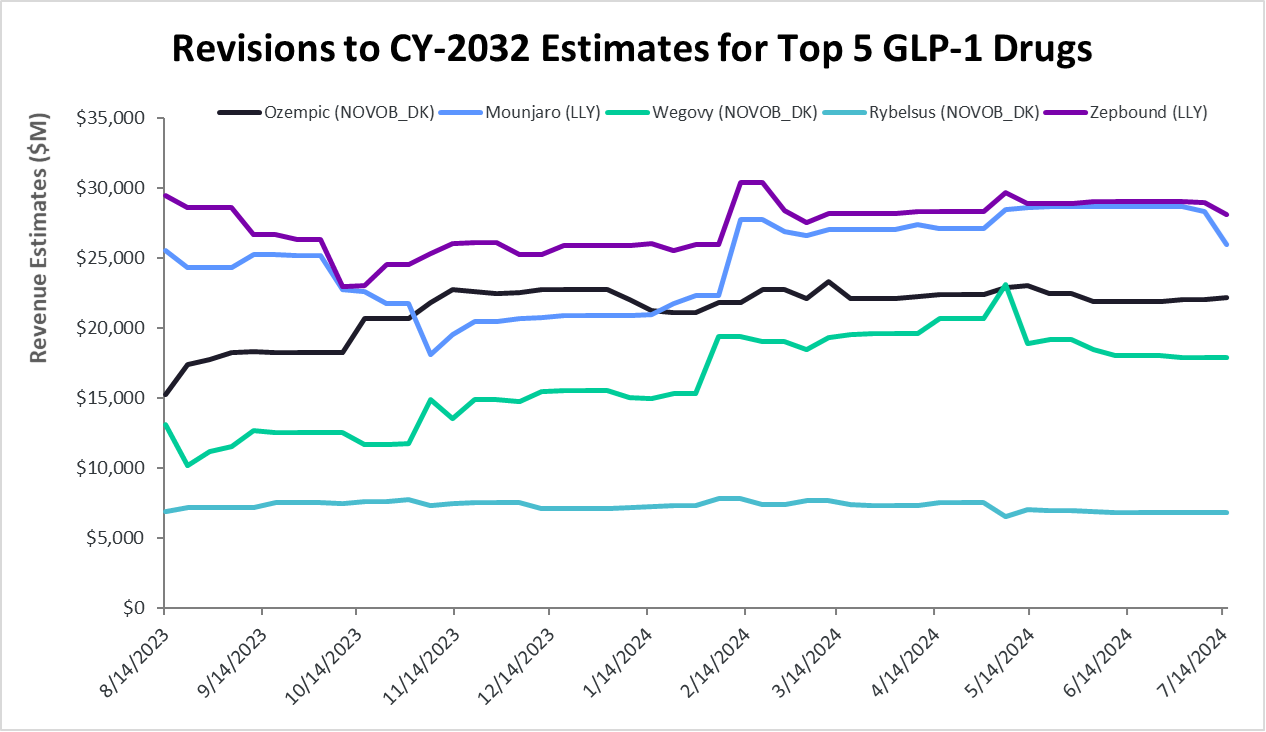
Figure 2: Revisions to 2032 Estimates for Five Leading GLP-1 Drugs
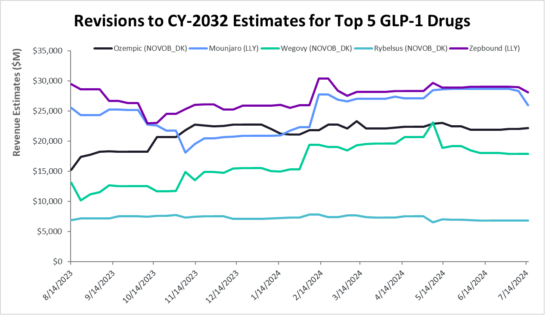
Source: Visible Alpha consensus (July 19, 2023)
Key Takeaways
- Visible Alpha consensus revenue projections of marketed GLP-1-based drugs remain steady since the release of our last GLP-1 monitor. Combined revenues for GLP-1 receptor agonists and related classes of drugs are projected to reach $174.7 billion by 2035 (our previous GLP-1 Monitor projected 2032 estimates at $164 billion, the updated GLP-1 Monitor projects 2032 revenues at $165.3 billion). Consensus revenue forecasts for each of the top 5 marketed drugs (Mounjaro, Zepbound, Ozempic, Wegovy, and Rybelsus) are in keeping with trends in our previous GLP-1 Monitor. A notable difference is that the 2035 projected revenue for Novo Nordisk’s (NVO) Phase 3 drug candidate CagriSema has jumped from $29 billion to almost $38 billion in this GLP-1 Monitor update.
- The next generation of GLP-1 competitor drug candidates, with novel mechanisms of action or improved drug profiles, are maturing as they progress through clinical development toward potential market approval. Several of these new competitors are in Phase 3 studies and are projected to enter the market by 2025 or shortly thereafter. The leading Phase 3 GLP-1-based drug candidates belong to Novo Nordisk (NVO) and Eli Lilly (LLY) – potentially maintaining the NVO – LLY dominance for the near future. Based on Visible Alpha consensus, the highest revenue projection amongst the leading Phase 3 drug candidates is for NVO’s CagriSema.
- Novel mechanisms of action in the future pipeline go beyond GLP-1 receptor agonists (NVO’s semaglutides) and dual GLP-1/GIP receptor agonists (LLY’s tirzepatides). The novel mechanisms of action include new metabolic pathways targeting amylin receptor agonism, glucagon receptor agonism, GLP-2 receptor agonists, and GIP receptor antagonists (not agonists) amongst others. These new metabolic pathway-targeting drugs are mostly in combination with a GLP-1 receptor agonist as a foundation.
|
Competitive Landscape of Emerging GLP-1 Drug Candidates
The next wave of GLP-1-based drug candidates, currently in Phase 3, will be dominated by NVO and LLY, further consolidating their dominance (Table 2). These include NVO’s CagriSema (GLP-1 receptor agonist + amylin receptor agonist) and LLY’s orfoglipiron (oral GLP-1 receptor agonist) and retatrutide (GLP-1 receptor agonist + GIP receptor agonist + glucagon receptor agonist).
NVO and LLY to maintain dominance for now; could face challenges from developing pipeline
The top 5 leading GLP-1 drugs currently on the market are owned by Novo Nordisk (NYSE: NVO) and Eli Lilly (NYSE: LLY). Together, these two companies captured 98.1% of total GLP-1 drug revenues in 2023, amounting to $37.2 billion out of approximately $37.9 billion. Looking ahead to 2033, based on Visible Alpha consensus, NVO and LLY are expected to maintain their dominant position, accounting for 89.9% of all GLP-1 drug revenues. By 2033, the combined NVO and LLY GLP-1-based drug revenues are expected to reach $152.5 billion. Overall, GLP-1-based drugs are forecasted to generate $169.7 billion in revenue in 2033.
However, the projected 2033 dynamics could shift if any of the competing Phase 3 or Phase 2 drug candidates are approved and show superiority over the current GLP-1 portfolios of NVO and LLY, either on the market or in development pipelines. Other companies advancing next-generation drugs in Phase 3/Phase 2 stages, aside from NVO and LLY, include (Table 2 and Table 3):
- Zealand Pharma (CPH: ZEAL)
- Pfizer (NYSE: PFE)
- Amgen (NASDAQ: AMGN)
- Structure Therapeutics (NASDAQ: GPCR)
- Altimmune (NASDAQ: ALT)
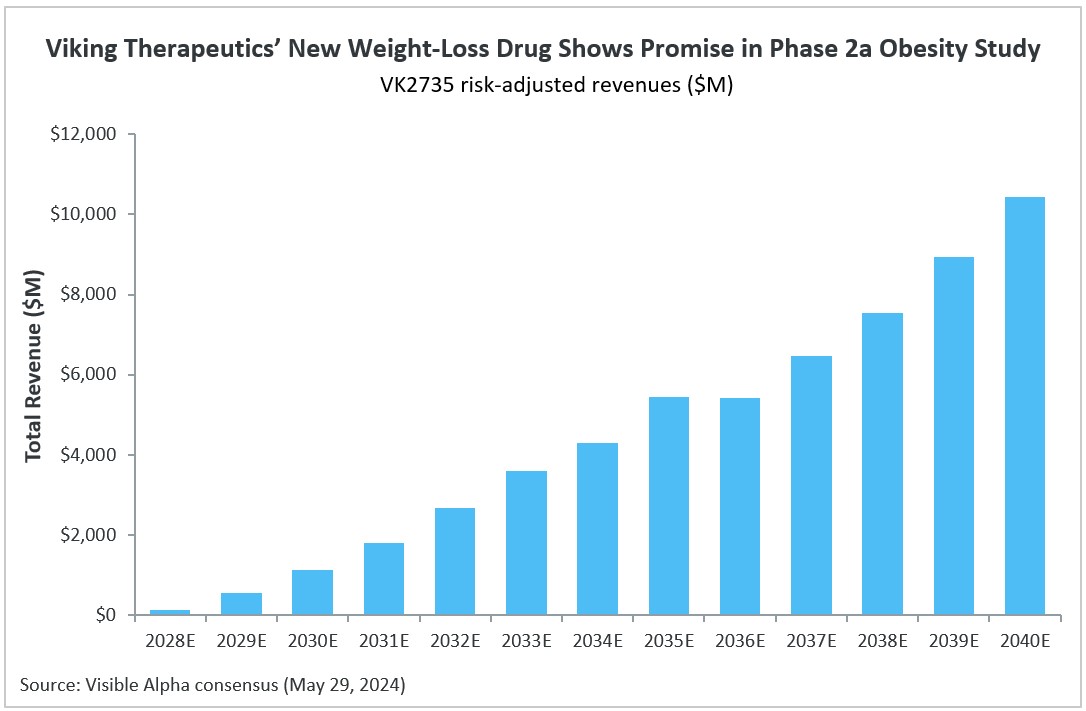
- Viking Therapeutics (NASDAQ: VKTX)
- Merck (NYSE: MRK)
- Roche (SIX:ROG)/Genentech
- AstraZeneca (NASDAQ, LSE: AZN)
- Terns Pharma (NASDAQ: TERN)
Notably, the only Phase 3 candidate outside of LLY and NVO’s offerings is ZEAL and Boehringer Ingelheim’s (private) Survodutide.
Novel Mechanisms & Improved Drug Profiles in the Pipeline
While GLP-1 receptor agonism and dual GLP-1 and GIP receptor agonists continue to be successful, new combinatorial approaches are being developed. These engage additional metabolic pathways that could potentially generate improved glucose regulation, greater weight loss, increased durability of weight loss, and improved tolerability/safety.
These innovative yet related metabolic pathway approaches target novel biology and mechanisms distinct from GLP-1 receptor agonism (semaglutides – Ozempic, Wegovy, Rybelsus) and dual GLP-1 and GIP receptor agonists (tirzepatides – Mounjaro, Zepbound).
GLP-1 and GLP-1 pathway-related drug candidates with new mechanisms of action include:
- GLP-1 receptor agonist & amylin receptor agonist (cagrilintide) – CagriSema (NVO), amycretin (NVO)
- GLP-1 receptor agonist & glucagon receptor agonist – mazdutide /LY3305677 (LLY), pemvidutide/ALT-801 (ALT), survodutide/BI-456906 (Boehringer Ingelheim & ZEAL), efinopegdutide/MK-6024 (MRK),
- GLP-1 receptor agonists & insulin analog – Icosema/NN1535 (NVO)
- GLP-1 receptor agonist & glucagon receptor agonist & GIP receptor agonist – retatrutide (LLY), AZD9550 (AZN)
- GLP-1 receptor agonists & GIP receptor antagonist – AMG-133/maridebart + cafraglutide (AMGN)
- GLP-1 receptor agonists & dapagliflozin, a SGLT2 inhibitor – SemaDapa (NVO)
- GLP-1 receptor agonists & GLP-2 receptor agonists – dapiglutide (ZEAL)
- Amylin receptor agonist (no GLP-1) – petrelintide, (ZEAL), AZD6234 (AZN)
Besides mechanism of action, innovative approaches to improve drug profile include – oral versions of current injections or novel oral formulation drug candidates, long-acting and less frequently dosed drug candidates, but with efficacy that is comparable or better to the currently approved drugs. These include oral GLP-1 receptor agonists such as PF-06882961/danuglipron by Pfizer, which is a once-daily oral pill in Phase 2, or NVO’s NN9650 that is a GLP-1 receptor agonist that is a once monthly dual GLP1/GIP receptor agonists.
The novel mechanisms and new formulations (oral, long-acting) aim to differentiate from the competition by improving the drug profile via one metric or the other – efficacy, safety, tolerability, and/or clinical outcomes across the board or within distinct patient sub-populations.
Table 1: Approved GLP-1-based drugs:
Semaglutides (Ozempic, Rybelsus, Wegovy) are GLP-1 receptor agonists; tirzepatides (Mounjaro and Zepbound) are GLP-1 receptors, and GIP receptor agonists. Liraglutide, dulaglutide, and exenatide are first generation GLP-1 based drugs
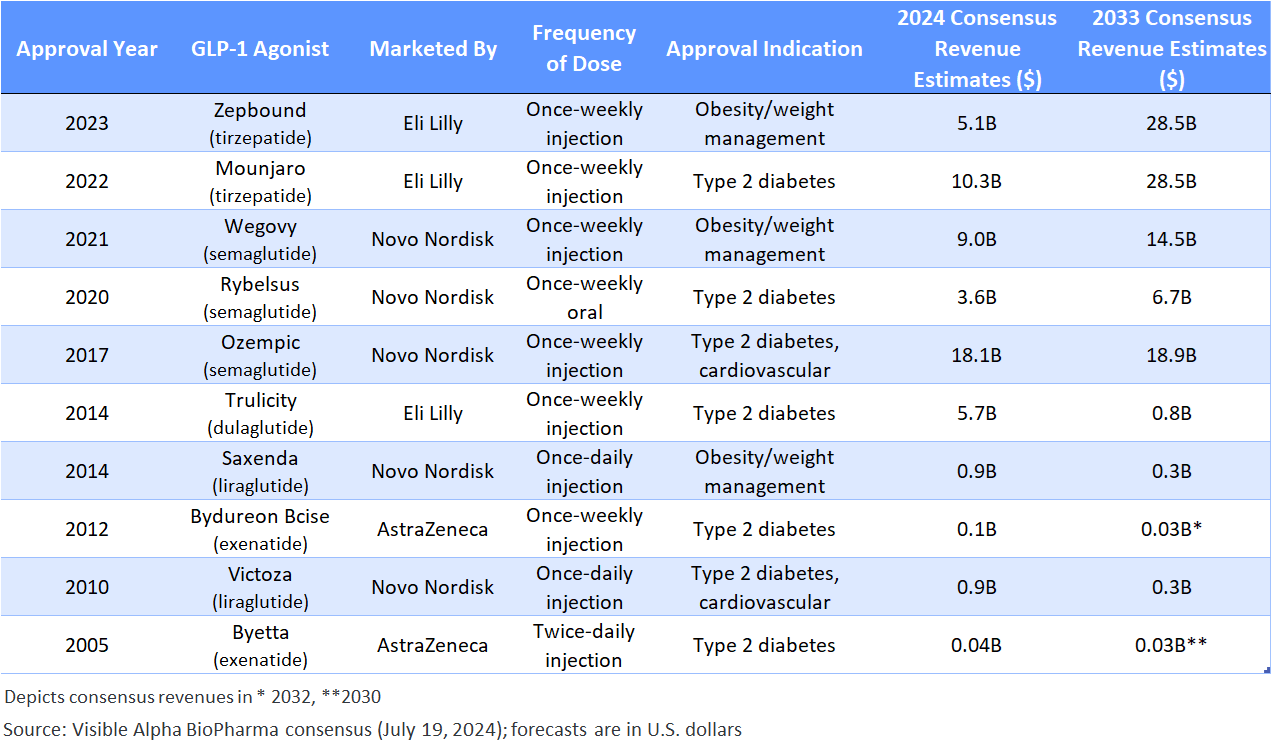
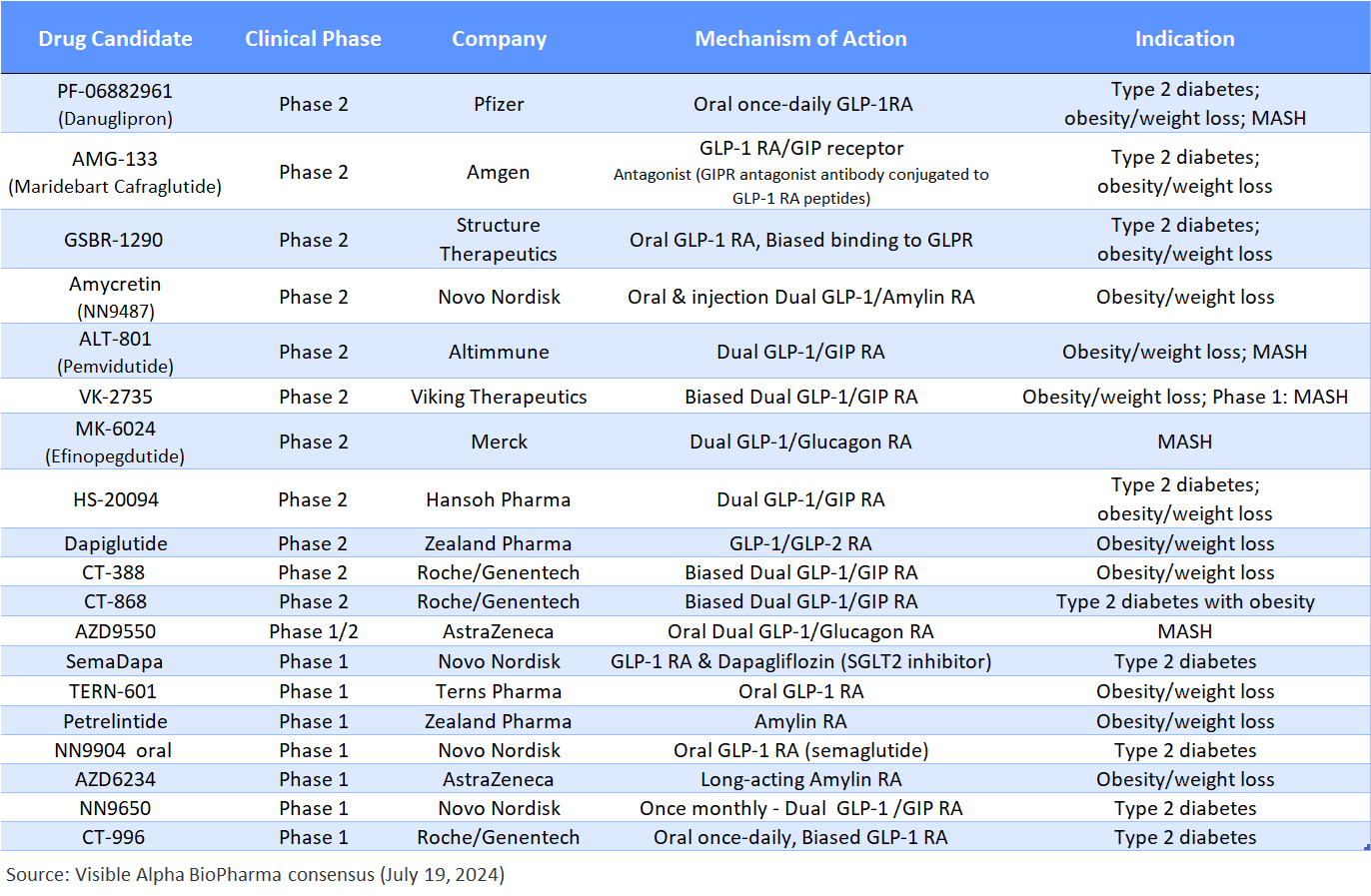
Table 2: GLP-1-based drug candidates in Phase 3 studies – NVO and LLY dominate the Phase 3 pipeline:
Emerging GLP-1-based drug candidates with a novel mechanism of action or an improved drug profile (e.g., oral versus injectable) are designed to compete with the current market leaders, if approved.

Table 3: GLP-1-based pipeline of drug candidates in Phase 2 and Phase 1 studies:
Unlike the Phase 3 pipeline, the Phase 2 pipeline of drugs is not dominated by LLY and NVO. Note that petrelintide (ZEAL) and AZD6234 (AZN) are amylin receptor agonists that are not GLP-1 pathway-based. AMG-133 (AMGN) is a GIP receptor antagonist (not agonist). MASH is metabolic dysfunction-associated steatohepatitis – a similar disease to the prior term, NASH.
Notable Novel Mechanisms of Action of GLP-1 and GLP-1-related Pathways in Clinical Development
GIP receptor antagonism (not agonism): GIP receptor agonism as occurs in Mounjaro and Zepbound, works by regulating energy balance through cell-surface receptor signaling in the brain and adipose tissue and by enhancing lipid storage and acts on the central nervous system to lower food intake. A notable and differentiating mechanism of action in the developing pipeline includes Amgen’s AMG-133 which is a GLP-1 receptor agonist and GIP receptor antagonist. AMG-133 is a bispecific construct engineered by conjugating a monoclonal anti-human GIP receptor antagonist antibody to two GLP-1 analogue agonist peptides using amino acid linkers. Pre-clinical and early clinical data has demonstrated that antagonizing GIP receptors (in combination with GLP-1 agonism) leads to improved weight loss and improvements in metabolic metrics.
Biased GLP-1 receptor agonism, the next generation of GLP-1 receptor agonists: Conventional GLP-1 receptor agonism involves full agonism by engaging multiple intracellular signaling pathways. In contrast, selective or biased agonism by engaging select intracellular signaling pathways results in distinct physiological pathways. Biased agonism of the GLP-1 receptor can enhance glucose regulatory efficacy by avoiding GLP-1 receptor desensitization and downregulation, by reduced β-arrestin pathway engagement and intracellular signaling. This effect can be achieved without disturbing the intracellular cAMP signaling pathway that leads to the multiple beneficial metabolic effects associated with GLP-1 receptor agonism. Companies developing biased GLP-1 receptor agonists include Structure Therapeutics (GSBR-1290 in Phase 2) and Roche/Genentech (RG6641/CT-868, RG6640/CT-388, RG6652/CT-996 in Phase 2 and Phase 1).
Amylin receptor agonism: Approaches that target the amylin receptor are differentiated from GLP-1-based therapies. Amylin is co-secreted with insulin and assists in glucose control. It inhibits glucagon secretion, delays gastric emptying, and signals satiety, suppressing the intake of food. Amylin is released from the pancreas and induces feelings of satiety, in contrast, GLP-1 is secreted by the gut and primarily reduces appetite. Early data suggests that amylin may have a more tolerable safety profile, however, larger clinical trials are needed to confirm. Amylin analogs could capture a market population that does not tolerate GLP-1-based drugs. We are aware of two drug candidates that target amylin exclusively – petrelintide (ZEAL) and AZD6234 (AZN), both in Phase 1 studies for obesity/weight loss. In addition, amylin receptor agonists in combination with GLP-1 receptor agonists are in clinical studies – CagriSema (NVO) in Phase 3 for type 2 diabetes and obesity/weight loss, amycretin (NVO) in Phase 2 for obesity/weight loss and NN9487 (NVO) an oral GLP-1 plus amylin analog in Phase 1 studies for obesity/weight loss.
Glucagon receptor agonists: Glucagon is a peptide hormone secreted from the alpha cells of the pancreatic islets of Langerhans. Glucagon plays important roles in glucose, amino acid, and fat metabolism and may also regulate appetite and energy expenditure. Glucagon regulates liver lipid metabolism by stimulating lipolysis and fatty acid oxidation and inhibiting lipogenesis, thereby reducing liver storage of fat. The combination of a GLP-1 receptor agonist and a glucagon receptor agonist has utility in obesity/weight loss via GLP-1 receptor agonism and reduction in liver fat via glucagon receptor agonism for fatty liver disease or MASH (metabolic dysfunction-associated steatohepatitis). MASH is the new term for nonalcoholic steatohepatitis (NASH).
Next Generation GLP-1 Drug Candidates Nearing Approval: NVO’s Cagrisema Stands Out
Based on Visible Alpha consensus revenue projections, NVO’s Cagrisema, and LLY’s Retatrutide and Orfoglipiron are the leading candidates to compete with the currently marketed GLP-1 drugs in the near term. All three could potentially be on the market in 2025.
Cagrisema, in Phase 3 for type 2 diabetes and obesity/weight loss, has the highest market potential. CagriSema is a combination of a GLP-1 receptor agonist (semaglutide) plus an amylin receptor agonist (Cagrilintide). Based on Visible Alpha consensus, by the year 2035, CagriSema’s risk-adjusted revenues are expected to top $37.9 billion, which is significantly more than LLY’s Retatrutide and Orfoglipiron revenue estimates. Risk-adjusted revenues for Retatrutide and Orfoglipiron in 2035 are $9.3 billion and $14.6 billion, respectively.
Notably, Cagrisema risk-adjusted revenue projections are even more than the revenue projections for the currently approved blockbuster GLP-1 drugs – Ozempic, Wegovy, Mounjaro, or Zepbound.
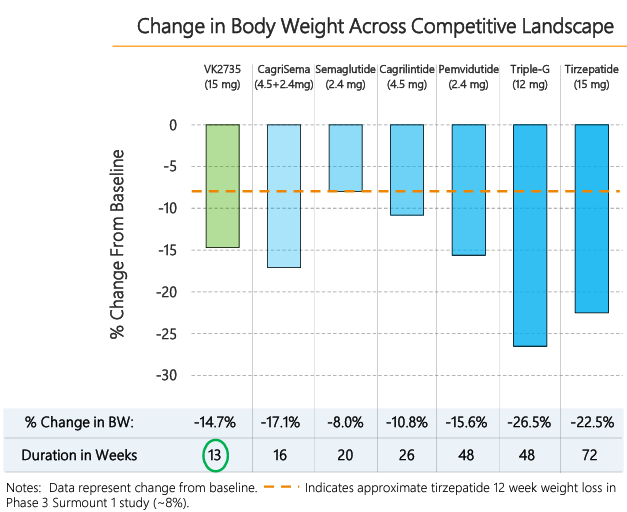
Figure 3: Comparing the Phase 3 GLP-1-based drug candidates:
Of the next-generation GLP-1-based drug candidates, currently in Phase 3, NVO’s Cagrisema, if approved, could reach almost $38 billion in risk-adjusted revenues by 2035. In comparison, risk-adjusted revenues for LLY’s Retatrutide and Orfoglipiron are dramatically lower.
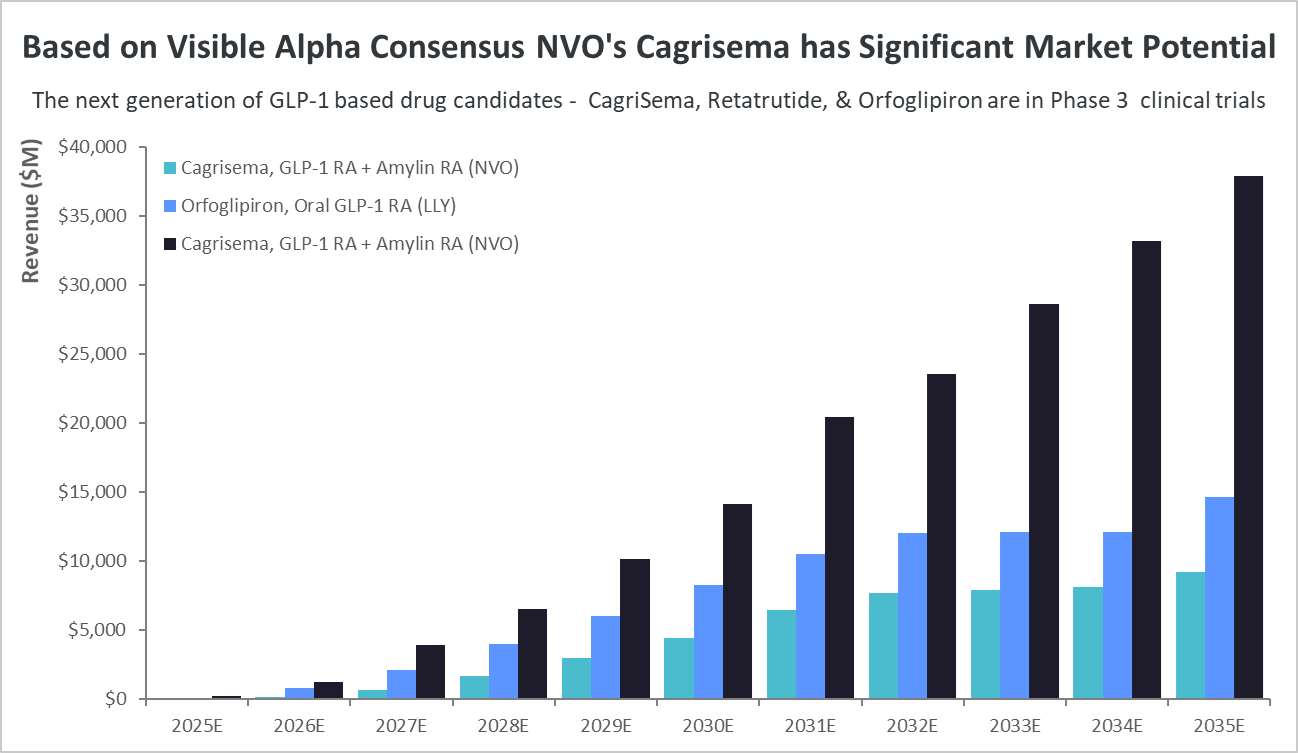
Source: Visible Alpha consensus (July 19, 2024)
Wegovy’s Cardiovascular Benefit Label Expansion Approved by the FDA
Wegovy is the first weight loss drug that is also approved to help prevent life-threatening cardiovascular events in adults with cardiovascular disease and either obesity or overweight (not diabetic). The FDA announced its decision in March 2024.
This will compel health insurance agencies currently not covering weight loss drugs to cover Wegovy if the patient is at risk of cardiovascular disease, which is often the case in obese/overweight patients. Medicare is prohibited by law to cover drugs for obesity/weight loss. However, the FDA’s extension of the Wegovy label to include cardiovascular benefits has changed Medicare’s view.
The FDA’s decision was based on a multicenter, double-blind, randomized, placebo-controlled, outcomes study (SELECT Trial) conducted by NVO and published in the New England Journal of Medicine in November 2023 (Lincoff et. al., 2023 NEJM. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes). The trial began in 2018 and enrolled 17,604 patients across 41 countries.
This is an advantage for Wegovy over its obesity/weight loss competitor Zepbound (LLY) since applicable Medicare patients can be reimbursed and avoid the steep cost (approximately $1,300 per month). Data suggests that the tirzepatides (Zepbound) may also offer cardiovascular in the obese non-diabetic population similar to semaglutides (Wegovy), however a completed study on cardiovascular risk-reduction has not been reported by LLY to date.
Effect of Wegovy in reducing cardiovascular risk is independent of weight loss & glucose control
At the recent American Diabetes Association meeting (June 21-24, 202) where data from the SELECT trial evaluating Wegovy’s cardiovascular benefits were presented, an area of much discussion was Wegovy’s beneficial effect on cardiovascular metrics independent of weight loss or glucose regulation. Reduction in inflammation is a factor in cardiovascular benefit. It is likely that along with glycemic, dyslipidemia, and blood pressure control, reduction in inflammation contributes significantly to cardiovascular benefit. Inflammation was measured by the high-sensitivity C-reactive protein (hs-CRP) test. This data confirmed that the semaglutide class (Wegovy) reduces inflammation, contributing to cardiovascular benefits independent of weight loss or glucose regulation. It remains to be seen if tirzepatides (Zepbound) will also have the same effect on inflammation and cardiovascular benefit as the semaglutides (Wegovy).
Conclusion
Next-generation GLP-1-based drug candidates with novel mechanisms of action are currently in Phase 3 trials and are projected to be approved in 2025. The next generation of drug candidates targeting GLP-1-related metabolic pathways in combination with GLP-1 receptor agonism harness complementary metabolic pathways that have the potential to improve glucose regulation, increase weight loss, and reduce cardiovascular risk with improved safety and tolerability. In addition, GLP-1 receptor agonist drug candidates with superior efficacy and an improved drug profile (biased agonism, long-acting or oral formulations) are also in development. The leading next-generation drug candidates in Phase 3, slated for approval in 2025, belong to either NVO or LLY, thus extending dominance over the competition. Based on Visible Alpha consensus, CagriSema (NVO) has the largest market potential of the next-generation GLP-1-based drug candidates currently in Phase 3.
The reduction in cardiovascular risk with GLP-1-based drugs for obesity (so far demonstrated with semaglutides) bodes well for further market expansion given medicare coverage and private insurance coverage of obesity/weight loss drugs.
As Big Pharma companies compete in the GLP-1 space, we anticipate partnering/acquisition transactions to continue to bolster Big Pharma pipelines with next-generation GLP-1 pathways and related drugs. Several biotechs with novel and promising GLP-1 and GLP-1 related pathway drug candidates in Phase 1 and 2 may be ripe for Big Pharma partnering/acquisition.